






Project on Sickle Cell Anaemia Class 12 Biology
Introduction
Sickle Cell Anaemia (SCA) is a autosomal recessive genetic blood disorder characterized by red blood cells that assume an abnormal, rigid, sickle shape.
This condition can lead to various complications, including pain, infection, and organ damage. Understanding the genetic basis, symptoms, complications, and management of SCA is crucial for improving patient outcomes and developing effective treatments.
Genetic Basis of Sickle Cell Anaemia
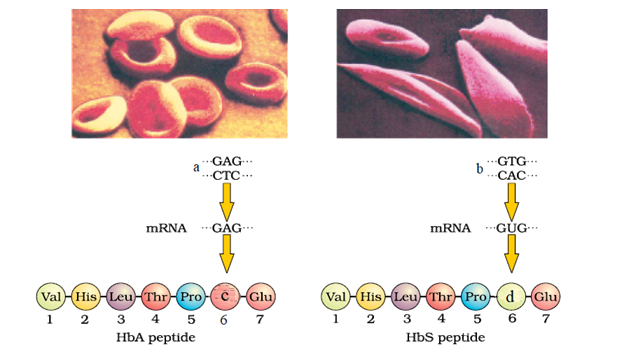
Sickle Cell Anaemia is caused by a mutation in the HBB gene, which encodes the beta-globin subunit of haemoglobin.
The mutation results in the substitution of Valine amino acid for glutamic amino acid at the sixth position of the beta-globin chain, forming haemoglobin S (HbS). Individuals with SCA inherit two copies of the HbS gene, one from each parent.
Those with one HbS gene and one normal haemoglobin gene (HbA) have sickle cell trait (SCT) and typically do not exhibit symptoms of the disease.
Epidemiology
Sickle Cell Anaemia predominantly affects individuals of African, Mediterranean, Middle Eastern, and Indian ancestry.
In the United States, it is most common among African Americans, with approximately 1 in 365 African American births affected by SCA. Globally, around 300,000 infants are born with SCA each year, with the highest prevalence in sub-Saharan Africa.
Pathophysiology of SCA
The abnormal haemoglobin in SCA leads to the sickling of red blood cells under low oxygen conditions.
These sickle-shaped cells are rigid and can obstruct blood flow in the microcirculation, leading to ischemic tissue damage and pain. Additionally, sickle cells have a shorter lifespan (10-20 days) compared to normal red blood cells (120 days), resulting in chronic haemolytic Anaemia.
Symptoms of SCA
The clinical presentation of SCA varies widely among individuals. Common symptoms include:
1. Pain Crises: Episodes of severe pain, known as vaso-occlusive crises, occur when sickle cells block blood flow to bones and organs.
2. Anaemia: Chronic haemolytic Anaemia leads to fatigue, pallor, and shortness of breath.
3. Infections: Increased susceptibility to infections due to functional asplenia (spleen damage) and immune dysfunction.
4. Dactylitis: Pain and swelling in the hands and feet, often the first symptom in infants.
5. Acute Chest Syndrome: A severe complication characterized by chest pain, fever, and respiratory distress.
6. Stroke: Increased risk of ischemic stroke due to vaso-occlusion in cerebral vessels.
Complications related to SCA
Sickle Cell Anaemia can lead to numerous complications, some of which can be life-threatening:
1. Organ Damage: Chronic organ damage due to repeated vaso-occlusion, affecting the spleen, liver, kidneys, and lungs.
2. Pulmonary Hypertension: High blood pressure in the lungs, leading to heart failure.
3. Leg Ulcers: Non-healing sores on the lower legs due to poor blood flow.
4. Priapism: Painful, prolonged erection of the penis due to sickling in penile blood vessels.
5. Gallstones: Formation of gallstones due to increased breakdown of red blood cells.
Diagnosis of Sickle Cell Anaemia
Sickle Cell Anaemia is typically diagnosed through new-born screening programs, which use techniques such as:
1. Haemoglobin Electrophoresis: Identifies different types of haemoglobin in the blood.
2. High-Performance Liquid Chromatography (HPLC): Measures the amounts of different haemoglobins.
3. Genetic Testing: Detects mutations in the HBB gene.
Prenatal diagnosis can also be performed using amniocentesis or chorionic villus sampling.
Management of Sickle Cell Anaemia
Management of Sickle Cell Anaemia involves both preventive and therapeutic strategies:
1. Hydroxyurea: A medication that increases fetal haemoglobin (HbF) levels, reducing the frequency of pain crises and other complications.
2. Blood Transfusions: Regular transfusions to maintain normal haemoglobin levels and prevent complications such as stroke.
3. Pain Management: Use of analgesics, hydration, and oxygen therapy to manage pain crises.
4. Antibiotics and Vaccinations: Prophylactic antibiotics (e.g., penicillin) and vaccinations to prevent infections.
5. Bone Marrow Transplantation: The only curative treatment, though limited by donor availability and risks associated with the procedure.
Research and Advances
Recent research has focused on novel therapies and genetic approaches to treat Sickle Cell Anaemia:
1. Gene Therapy: Techniques to correct the defective HBB gene or introduce functional copies of the gene.
2. CRISPR-Cas9: Gene-editing technology to modify specific DNA sequences and potentially cure SCA.
3. New Drug Development: Investigating drugs that target different pathways involved in sickling and inflammation.
Psychosocial Impact
Living with Sickle Cell Anaemia poses significant psychosocial challenges. Patients may experience:
1. Chronic Pain and Fatigue: Affecting daily activities and quality of life.
2. Emotional Stress: Due to frequent hospitalizations and uncertainty about the future.
3. Social Stigma: Misunderstanding and discrimination related to the disease.
4. Financial Burden: High costs of medical care and potential loss of income due to illness.
Support from healthcare providers, family, and patient advocacy groups is crucial for managing these challenges.
Treatment of Sickle Cell Anaemia
Sickle Cell Anaemia (SCA) is a chronic condition that requires a multifaceted approach to treatment. The goals of treatment are to manage symptoms, prevent complications, and improve the quality of life for patients. Treatment strategies can be broadly categorized into pharmacological therapies, supportive care, and curative interventions.
Pharmacological Therapies
1. Hydroxyurea
-Mechanism: Hydroxyurea is a cytotoxic drug that increases the production of fetal hemoglobin (HbF), which reduces the sickling of red blood cells.
-Benefits: It decreases the frequency of pain crises, acute chest syndrome, and the need for blood transfusions. It also reduces mortality in SCA patients.
-Usage: It is typically administered orally on a daily basis. The dosage is adjusted based on the patient’s response and blood counts.
-Side Effects: Common side effects include bone marrow suppression, gastrointestinal disturbances, and an increased risk of infections.
2. L-Glutamine
-Mechanism: L-Glutamine is believed to reduce oxidative stress in sickle cells, thereby decreasing the frequency of pain crises.
-Benefits: It has been shown to reduce the frequency of hospital visits for pain and the occurrence of acute chest syndrome.
-Usage: It is taken orally twice a day.
-Side Effects: Side effects can include nausea, constipation, and abdominal pain.
3. Voxelotor
-Mechanism: Voxelotor binds to haemoglobin and increases its affinity for oxygen, which helps to prevent sickling of red blood cells.
-Benefits: It improves haemoglobin levels and reduces haemolysis.
-Usage: It is taken orally once daily.
-Side Effects: Common side effects include headache, diarrhea, and fatigue.
4. Crizanlizumab
-Mechanism: Crizanlizumab is a monoclonal antibody that targets P-selectin, a protein involved in the adhesion of sickle cells to the blood vessel walls.
-Benefits: It reduces the frequency of pain crises.
-Usage: Administered as an intravenous infusion once a month.
-Side Effects: Side effects can include nausea, back pain, and fever.
Supportive Care
1. Blood Transfusions
-Purpose: Blood transfusions are used to increase the number of normal red blood cells in the bloodstream, reducing Anaemia and the risk of complications such as stroke.
-Types: Simple transfusions or exchange transfusions, where a portion of the patient’s blood is replaced with donor blood.
-Risks: Potential risks include iron overload, alloimmunization (immune reaction to transfused blood), and infections.
2. Pain Management
-Medications: Analgesics such as acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDs), and opioids are used to manage pain crises.
-Non-pharmacological Methods: Techniques such as hydration, heat application, and cognitive-behavioural therapy can also help alleviate pain.
3. Antibiotics and Vaccinations
-Prophylactic Antibiotics: Penicillin is often prescribed to young children with SCA to prevent pneumococcal infections.
-Vaccinations: Patients should receive all routine vaccinations, including pneumococcal, Haemophilous influenza type b (Hib), meningococcal, and annual influenza vaccines.
4. Folic Acid Supplementation
-Purpose: Folic acid is given to support the increased production of red blood cells and prevent folate deficiency.
5. Management of Acute Complications
-Acute Chest Syndrome: Treated with oxygen therapy, antibiotics, bronchodilators, and blood transfusions.
-Splenic Sequestration: Rapidly treated with blood transfusions and sometimes splenectomy (surgical removal of the spleen).
-Stroke: Managed with exchange transfusions to reduce haemoglobin S levels and prevent recurrence.
Curative Interventions
1. Bone Marrow Transplantation (BMT)
-Mechanism: BMT replaces the patient’s defective hematopoietic stem cells with healthy stem cells from a compatible donor.
-Candidates: Best suited for children with severe SCA who have a matched sibling donor.
-Benefits: It can potentially cure SCA by enabling the production of normal red blood cells.
-Risks: Risks include graft-versus-host disease (GVHD), infections, and the complications associated with high-dose chemotherapy used to prepare the patient for the transplant.
2. Gene Therapy
-Mechanism: Experimental approaches include using viral vectors to introduce a normal beta-globin gene or using gene-editing technologies like CRISPR-Cas9 to correct the mutation in the patient’s hematopoietic stem cells.
-Status: Still under clinical investigation, with promising early results.
Lifestyle and Psychosocial Support
1. Education and Counselling
– Patients and families should receive education about the disease, treatment options, and strategies to manage symptoms and prevent complications.
2. Psychosocial Support
– Support from mental health professionals, social workers, and patient advocacy groups can help address the emotional and psychological challenges of living with SCA.
3. Healthy Lifestyle
– Encouraging a healthy diet, regular exercise, hydration, and avoiding triggers such as extreme temperatures and high altitudes can help manage the disease.
4. Regular Follow-up
– Routine follow-up with a haematologist and other specialists is essential for monitoring the disease and adjusting treatment as needed.
Conclusion
Sickle Cell Anaemia is a complex genetic disorder with significant health and social implications. Advances in research and treatment have improved the prognosis for many patients, but challenges remain. Continued efforts in education, early diagnosis, comprehensive care, and research into novel therapies are essential to further improve outcomes for individuals living with this disease.